



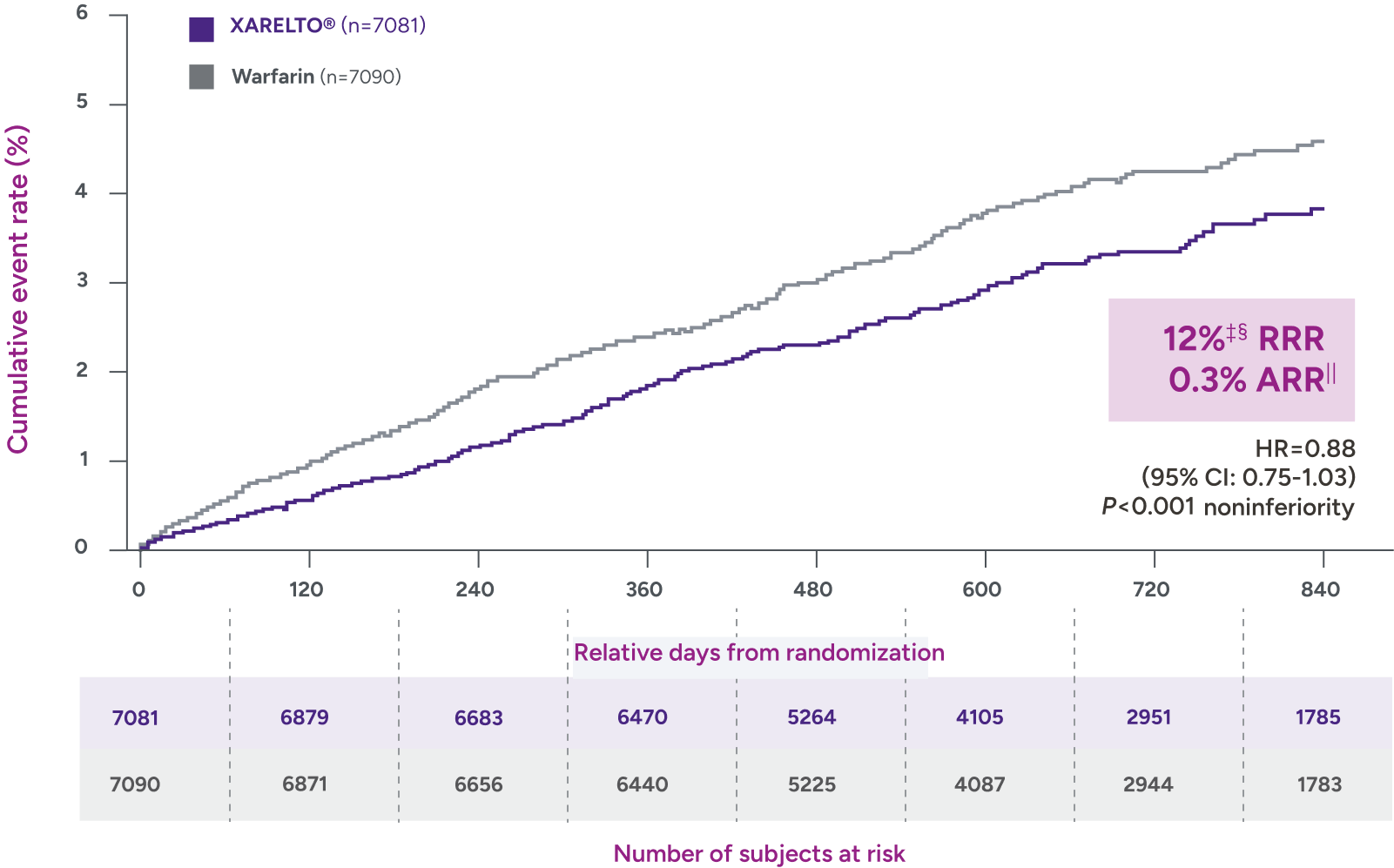
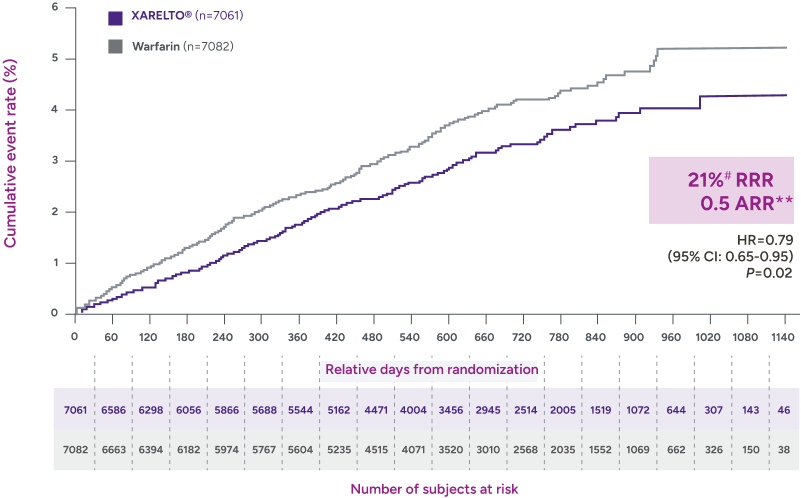


Rates of major bleeding vs warfarin‡


Study limitations:
- An EHR entry indicating initiation/use of an OAC does not guarantee a patient took it or allow for assessment of adherence or persistence, due to the lack of prescription claims data. Therefore, the analyses performed used an ITT approach
- Nonrandomized studies can be impacted by confounding, misclassification, and sampling biases
- Study findings were most generalizable to US population given the database used
- Time in therapeutic INR was not calculated for warfarin patients
- Additional analyses based on dose were not done due to the lack of INR data/therapeutic target range and the small proportion of rivaroxaban patients that were prescribed a low dose
- The database did not cover all institutions and therefore follow-up events may have been missed
- Investigation of the potential impact of socioeconomic status and patients’ actual incomes on study outcomes was not possible
- Reliable mortality data was not available
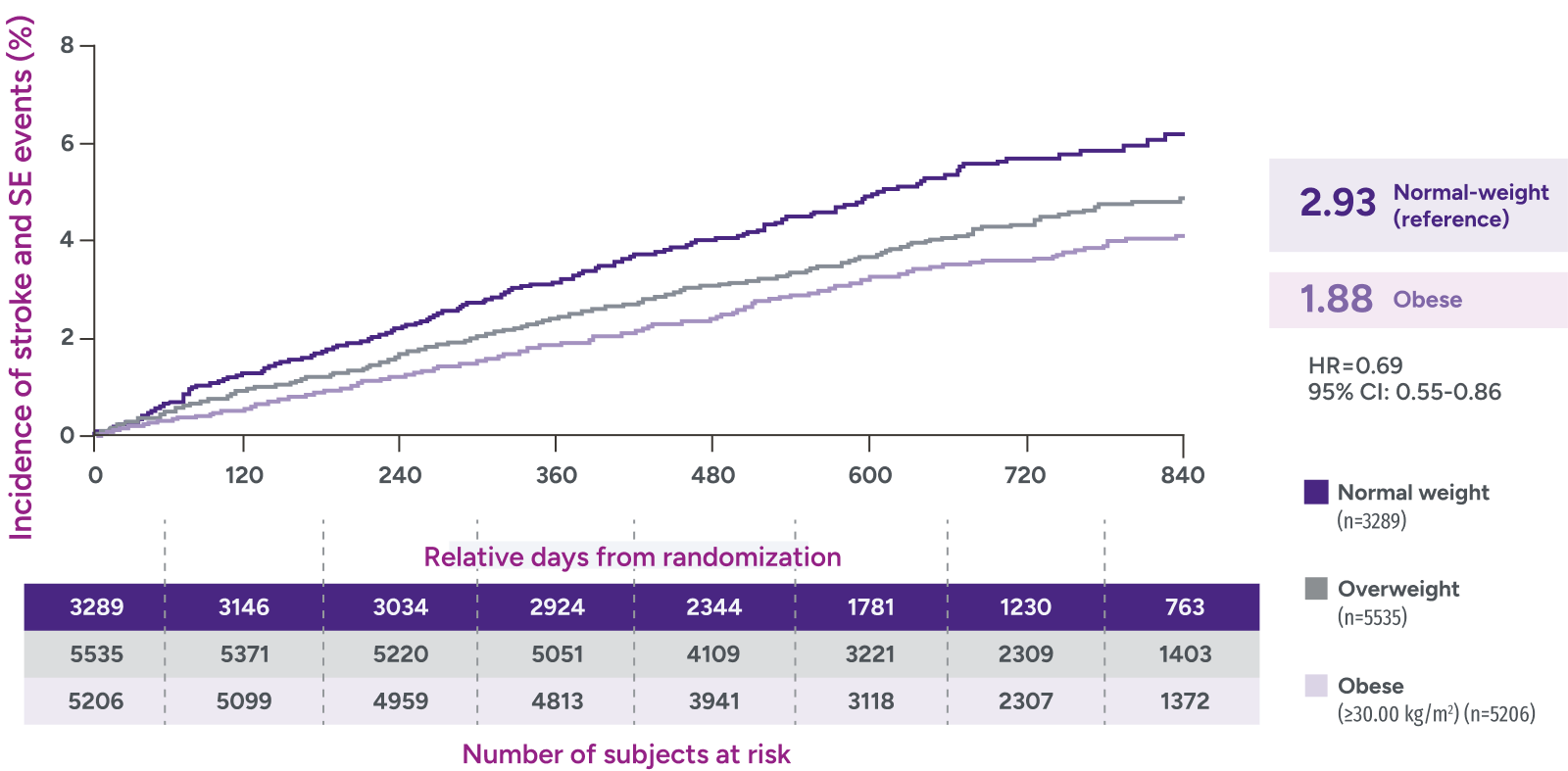
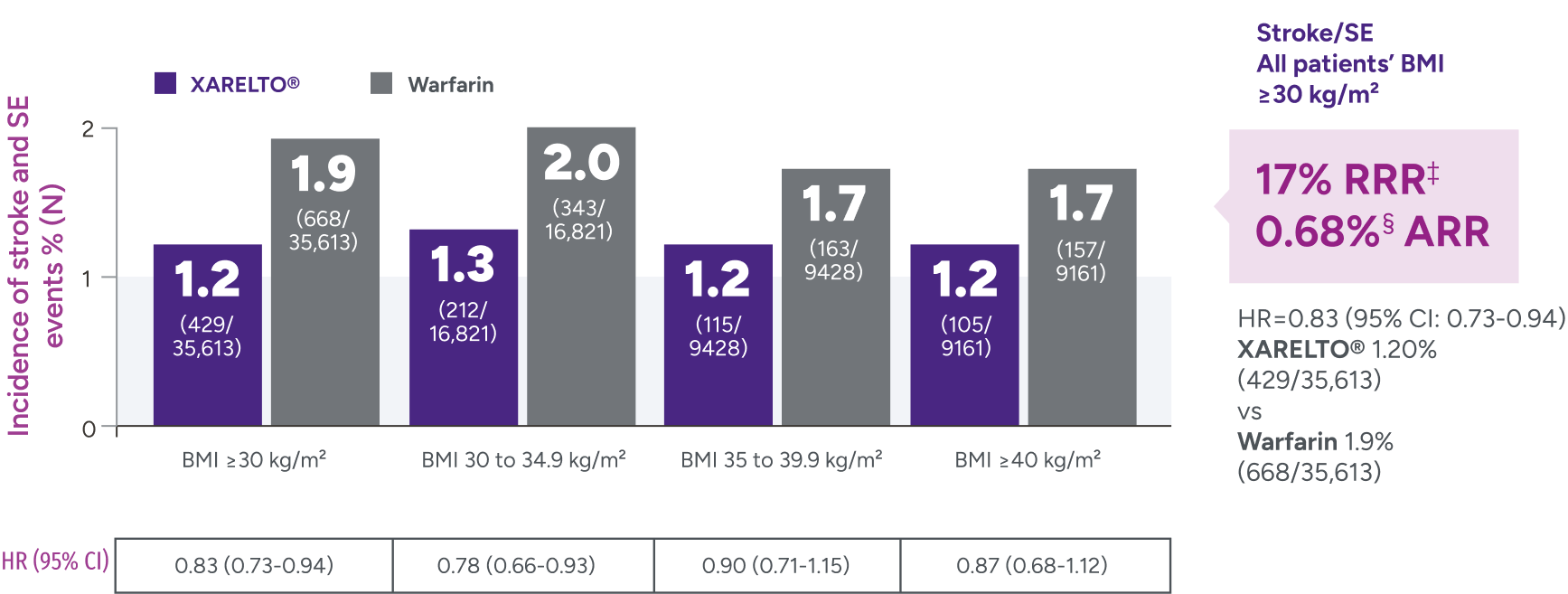
*Patients were identified as having obesity based on a BMI measurement of >30 kg/m2.
†Major bleeding was defined by the validated Cunningham algorithm.
‡Due to differences in study design, patient populations, definitions of outcomes, and data collection methods, the results of real-world studies are not intended for direct comparisons with clinical trials.
§RRR = (1 - 0.82) x 100% = 18%.
IIARR = 3.88% - 2.46% = 1.42%.
ARR = absolute risk reduction; BMI = body mass index; CI = confidence interval; EHR = electronic health record; HR = hazard ratio; INR = international normalized ratio; ITT = intent-to-treat; NVAF = nonvalvular atrial fibrillation; OAC = oral anticoagulant; RRR = relative risk reduction; RWE = real-world evidence; US = United States.


